
AI for Biochemistry
In biochemistry, understanding molecular structures and interactions is key but challenging due to the complexity of biological systems. Traditional methods for molecular modeling are slow, labor-intensive, and often struggle with data scalability and accuracy. These methods are also hindered by often incomplete or low-quality data, leading to inefficiencies in research and applications like drug development. Additionally, traditional computational approaches may not fully capture the dynamic nature of molecular interactions, limiting their adaptability and effectiveness across different molecular systems.
Basic Idea:
- Using Graph Neural Networks (GNNs) to model molecular structures as graphs, enhancing predictive accuracy and molecular insight.
- Utilizing large language models to interpret and generate molecular sequences, boosting innovation in molecule design.
- Developing methods to enhance dataset quality through informed synthetic data generation, improving model training and performance.
GIT-Mol: A Multi-modal Large Language Model for Molecular Science with Graph, Image, and Text
Paper Link: https://www.sciencedirect.com/science/article/pii/S0010482524001574
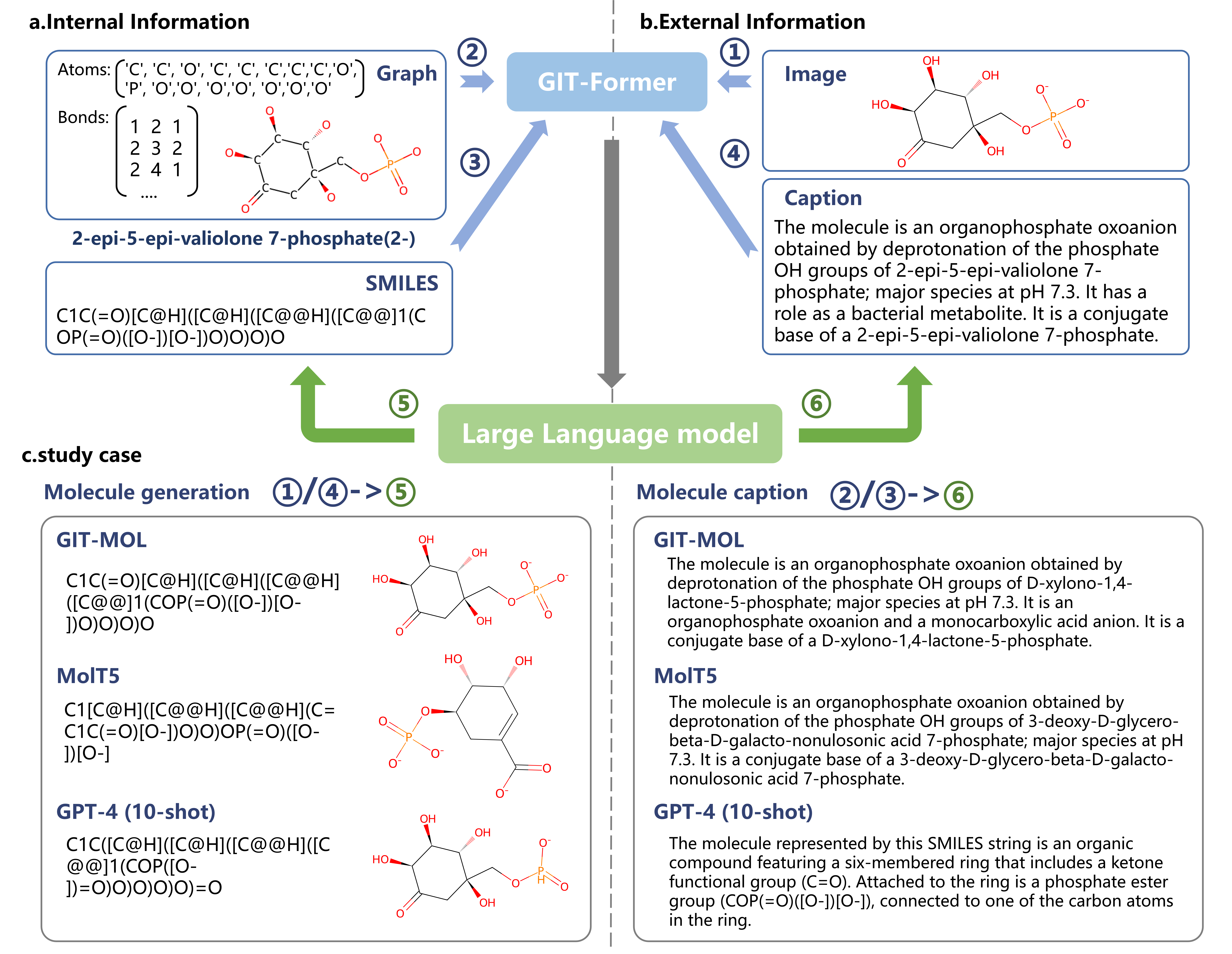
Large language models have made significant strides in natural language processing, enabling innovative applications in molecular science by processing textual representations of molecules. However, most existing language models cannot capture the rich information with complex molecular structures or images. In this paper, we introduce GIT-Mol, a multi-modal large language model that integrates the Graph, Image, and Text information. To facilitate the integration of multi-modal molecular data, we propose GIT-Former, a novel architecture that is capable of aligning all modalities into a unified latent space. We achieve a 5%-10% accuracy increase in properties prediction and a 20.2% boost in molecule generation validity compared to the baselines. With the any-to-language molecular translation strategy, our model has the potential to perform more downstream tasks, such as compound name recognition and chemical reaction prediction.
3D-Mol: A Novel Contrastive Learning Framework for Molecular Property Prediction with 3D Information
Paper Link: https://www.biorxiv.org/content/10.1101/2023.08.15.553467v1.abstract
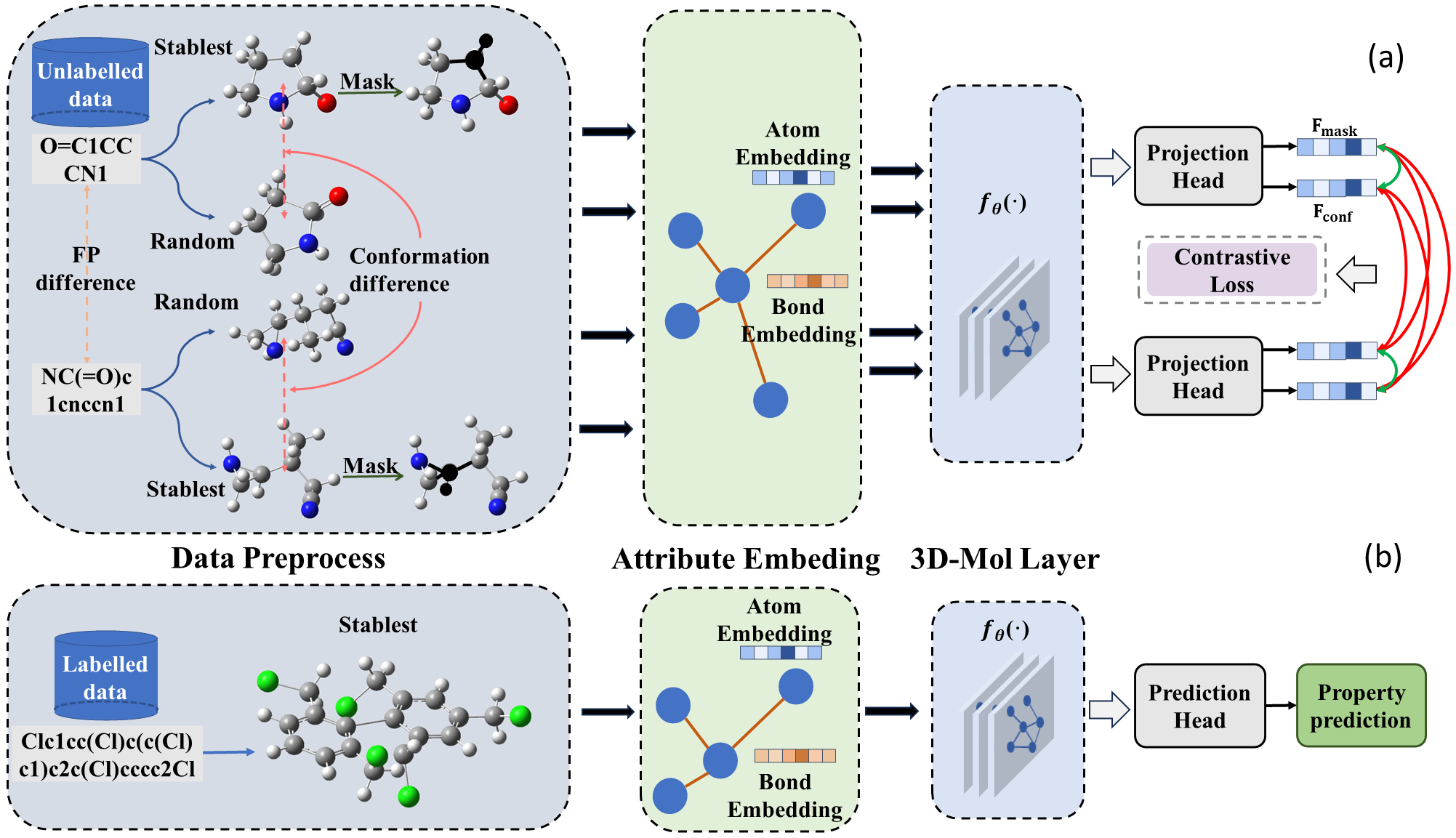
Molecular property prediction offers an effective and efficient approach for early screening and optimization of drug candidates. Although deep learning based methods have made notable progress, most existing works still do not fully utilize 3D spatial information. This can lead to a single molecular representation representing multiple actual molecules. To address these issues, we propose a novel 3D structure-based molecular modeling method named 3D-Mol. In order to accurately represent complete spatial structure, we design a novel encoder to extract 3D features by deconstructing the molecules into three geometric graphs. In addition, we use 20M unlabeled data to pretrain our model by contrastive learning. We consider conformations with the same topological structure as positive pairs and the opposites as negative pairs, while the weight is determined by the dissimilarity between the conformations. We compare 3D-Mol with various state-of-the-art(SOTA) baselines on 7 benchmarks and demonstrate our outstanding performance in 5 benchmarks.
A Self-feedback Knowledge Elicitation Approach for Chemical Reaction Predictions
Paper Link: https://arxiv.org/abs/2404.09606
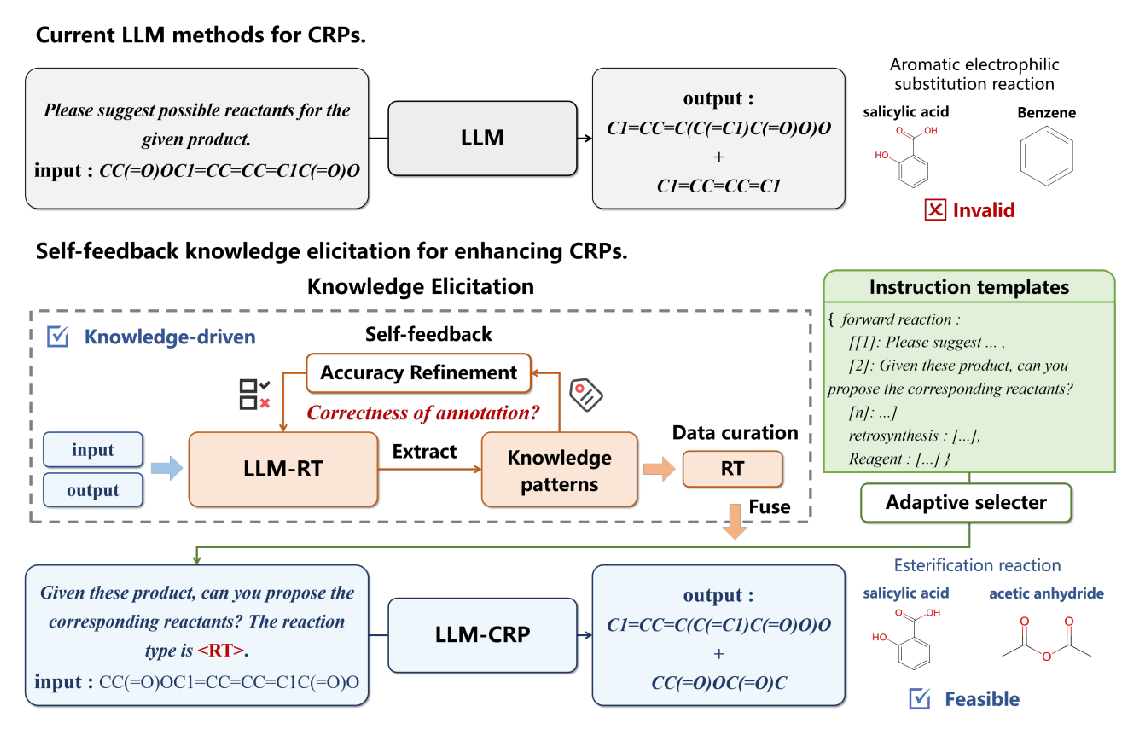
The task of chemical reaction predictions (CRPs) plays a pivotal role in advancing drug discovery and material science. However, its effectiveness is constrained by the vast and uncertain chemical reaction space and challenges in capturing reaction selectivity, particularly due to existing methods' limitations in exploiting the data's inherent knowledge. To address these challenges, we introduce a data-curated self-feedback knowledge elicitation approach. This method starts from iterative optimization of molecular representations and facilitates the extraction of knowledge on chemical reaction types (RTs). Then, we employ adaptive prompt learning to infuse the prior knowledge into the large language model (LLM). As a result, we achieve significant enhancements: a 14.2% increase in retrosynthesis prediction accuracy, a 74.2% rise in reagent prediction accuracy, and an expansion in the model's capability for handling multi-task chemical reactions. This research offers a novel paradigm for knowledge elicitation in scientific research and showcases the untapped potential of LLMs in CRPs.