
AI for Protein
Proteins play a pivotal role in the biological processes of living organisms, contributing to cell structure, functionality, signal transduction, and enzymatic reactions. Understanding proteins is crucial for various fields, including biochemistry, molecular biology, and medicine. In recent years, deep neural networks have achieved remarkable breakthroughs in the research of proteins, particularly in protein structure prediction such as the AlphaFold3. However, there remains a substantial gap in deciphering the intricate interplay and dependencies between protein structure and function. In addition, protein structures are inherently dynamic, undergoing complex conformational changes that are crucial for their function but challenging to capture with AI algorithms.
Basic Idea:
- Explore a comprehensive and explainable protein representation approach to bridge the gap from structure to function.
- Investigate the optimization or de novo design of proteins for specific functions, thereby opening up a reverse channel that connects function back to structure.
- Implement algorithms that can model the dynamic changes in protein structures over time.
DeepProSite: structure-aware protein binding site prediction using ESMFold and pretrained language model
Paper Link: https://academic.oup.com/bioinformatics/article/39/12/btad718/7453375
Identifying the functional sites of a protein, such as the binding sites of proteins, peptides, or other biological components, is crucial for understanding related biological processes and drug design. However, existing sequence-based methods have limited predictive accuracy, as they only consider sequence-adjacent contextual features and lack structural information. In this study, DeepProSite is presented as a new framework for identifying protein binding site that utilizes protein structure and sequence information. DeepProSite first generates protein structures from ESMFold and sequence representations from pretrained language models. It then uses Graph Transformer and formulates binding site predictions as graph node classifications. In predicting protein–protein/peptide binding sites, DeepProSite outperforms state-of-the-art sequence- and structure-based methods on most metrics. Moreover, DeepProSite maintains its performance when predicting unbound structures, in contrast to competing structure-based prediction methods. DeepProSite is also extended to the prediction of binding sites for nucleic acids and other ligands, verifying its generalization capability. Finally, an online server for predicting multiple types of residue is established as the implementation of the proposed DeepProSite. The datasets and source codes can be accessed at https://github.com/WeiLab-Biology/DeepProSite. The proposed DeepProSite can be accessed at https://inner.wei-group.net/DeepProSite/.
Running ahead of evolution—AI-based simulation for predicting future high-risk SARS-CoV-2 variants
Paper Link: https://journals.sagepub.com/doi/abs/10.1177/10943420231188077
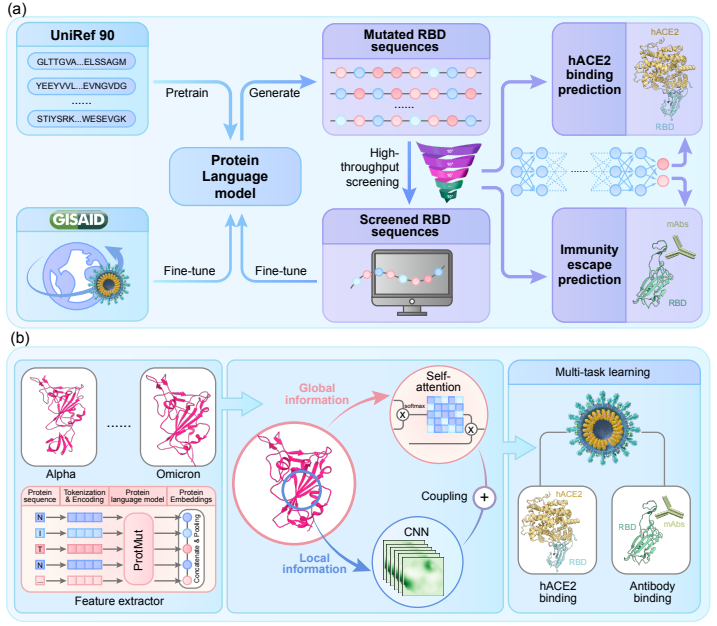
The never-ending emergence of SARS-CoV-2 variations of concern (VOCs) has challenged the whole world for pandemic control. In order to develop effective drugs and vaccines, one needs to efficiently simulate SARS-CoV-2 spike receptor-binding domain (RBD) mutations and identify high-risk variants. We pretrain a large protein language model with approximately 408 million protein sequences and construct a high-throughput screening for the prediction of binding affinity and antibody escape. As the first work on SARS-CoV-2 RBD mutation simulation, we successfully identify mutations in the RBD regions of 5 VOCs and can screen millions of potential variants in seconds. Our workflow scales to 4096 NPUs with 96.5% scalability and 493.9× speedup in mixed-precision computing, while achieving a peak performance of 366.8 PFLOPS (reaching 34.9% theoretical peak) on Pengcheng Cloudbrain-II. Our method paves the way for simulating coronavirus evolution in order to prepare for a future pandemic that will inevitably take place. Our models are released at https://github.com/ZhiweiNiepku/SARS-CoV-2_mutation_simulation to facilitate future related work.