
AI for Transcriptomics
High‐throughput sequencing technologies are now routinely being applied to a wide range of important topics in biology and medicine, including predictive modeling, drug discovery, biomarker discovery, medical decision making, and personalized medicine. The primary challenge in this domain is the diversity and complexity of the data, which makes manual extraction of features difficult and highly professional. Existing methods overfit the features of the current dataset, and the performance of the model is greatly degraded under real-world applications and noisy disturbances. Moreover, conventional scenarios tend to focus on omics data from a single source, ignoring the diversity and multiple sources of data.
Basic Idea:
- Using a multi-modalities integration framework to build new toolkits for cancer-related big data mining and knowledge discovery.
- Developing an efficient and generalized pre-train models for modeling heterogeneous data from different sources.
- Developing new biological computing platforms that leverage the proposed robust models and algorithms to solve new scientific problems and gain new insights into disease mechanisms, identify novel biomarkers of disease, and develop more effective therapies.
MetaSCDrug: Meta-Transfer Learning for Single-Cell-Level Drug Response Prediction from Transcriptome and Molecular Representations
Paper Link: https://www.biorxiv.org/content/10.1101/2024.04.25.591050.abstract
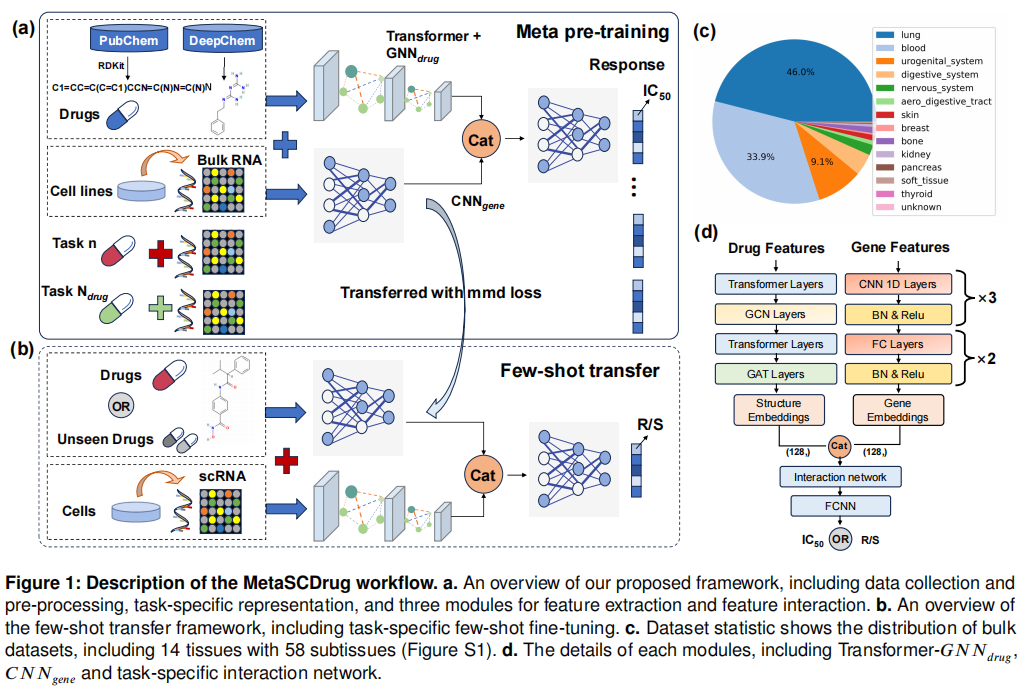
Analyzing the drug response at the cellular level is crucial for identifying biomarkers and understanding the mechanisms of resistance. Although studies on the drug response of individual cells can provide novel insights into tumor heterogeneity, pharmacogenomic data related to single-cell (SC) RNA sequencing is often limited. Transfer learning provides a promising approach to translate the knowledge of drug response from bulk cell lines to SC analysis, potentially providing an effective solution to this challenge. Previous studies often use data from single drug-cell lines to pre-train specific models and adapt the models on SC datasets, which lack pharmacogenomic information from other drugs and hinder model generalization. In this work, we introduce MetaSCDrug as a unified meta pre-training framework that integrates molecular information with transcriptomic data to simultaneously modeling cellular heterogeneity in response to multiple pre-trained drugs and generalize to unseen drugs. Our model requires only one pre-training session, followed by fine-tuning on multiple single-cell datasets by few-shot learning, achieving an average of 4.58% accuracy increase in drug response prediction compared to the baselines. Furthermore, our meta pre-training strategy effectively captures transcriptome heterogeneity in the generalization of unseen drugs, achieving a 20% improvement over the model without meta pre-training. Case studies of our framework highlight its capability to identify critical genes for resistance, providing a method for exploring drug action pathways and understanding resistance mechanisms.
Cytokine expression patterns: A single-cell RNA sequencing and machine learning based roadmap for cancer classification
Paper Link: https://www.sciencedirect.com/science/article/pii/S1476927124000136
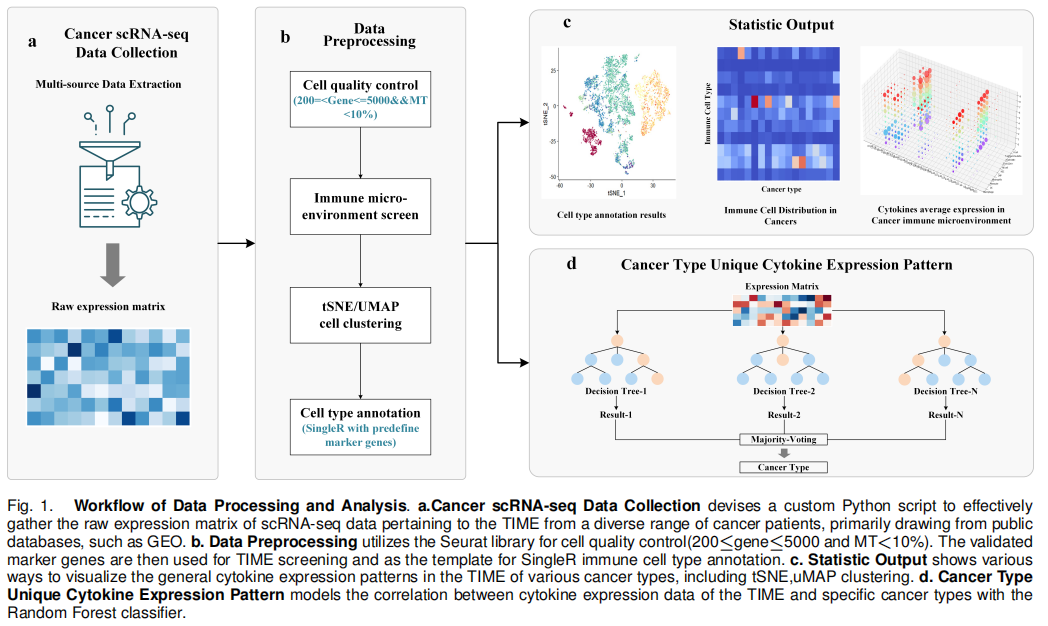
Cytokines are small protein molecules that exhibit potent immunoregulatory properties, which are known as the essential components of the tumor immune microenvironment (TIME). While some cytokines are known to be universally upregulated in TIME, the unique cytokine expression patterns have not been fully resolved in specific types of cancers. To address this challenge, we develop a TIME single-cell RNA sequencing (scRNA-seq) dataset, which is designed to study cytokine expression patterns for precise cancer classification. The dataset, including 39 cancers, is constructed by integrating 684 tumor scRNA-seq samples from multiple public repositories. After screening and processing, the dataset retains only the expression data of immune cells. With a machine learning classification model, unique cytokine expression patterns are identified for various cancer categories and pioneering applied to cancer classification with an accuracy rate of 78.01%. Our method will not only boost the understanding of cancer-type-specific immune modulations in TIME but also serve as a crucial reference for future diagnostic and therapeutic research in cancer immunity.
Self-supervised learning on millions of pre-mRNA sequences improves sequence-based RNA splicing prediction
Paper Link: https://academic.oup.com/bib/article/25/3/bbae163/7644137
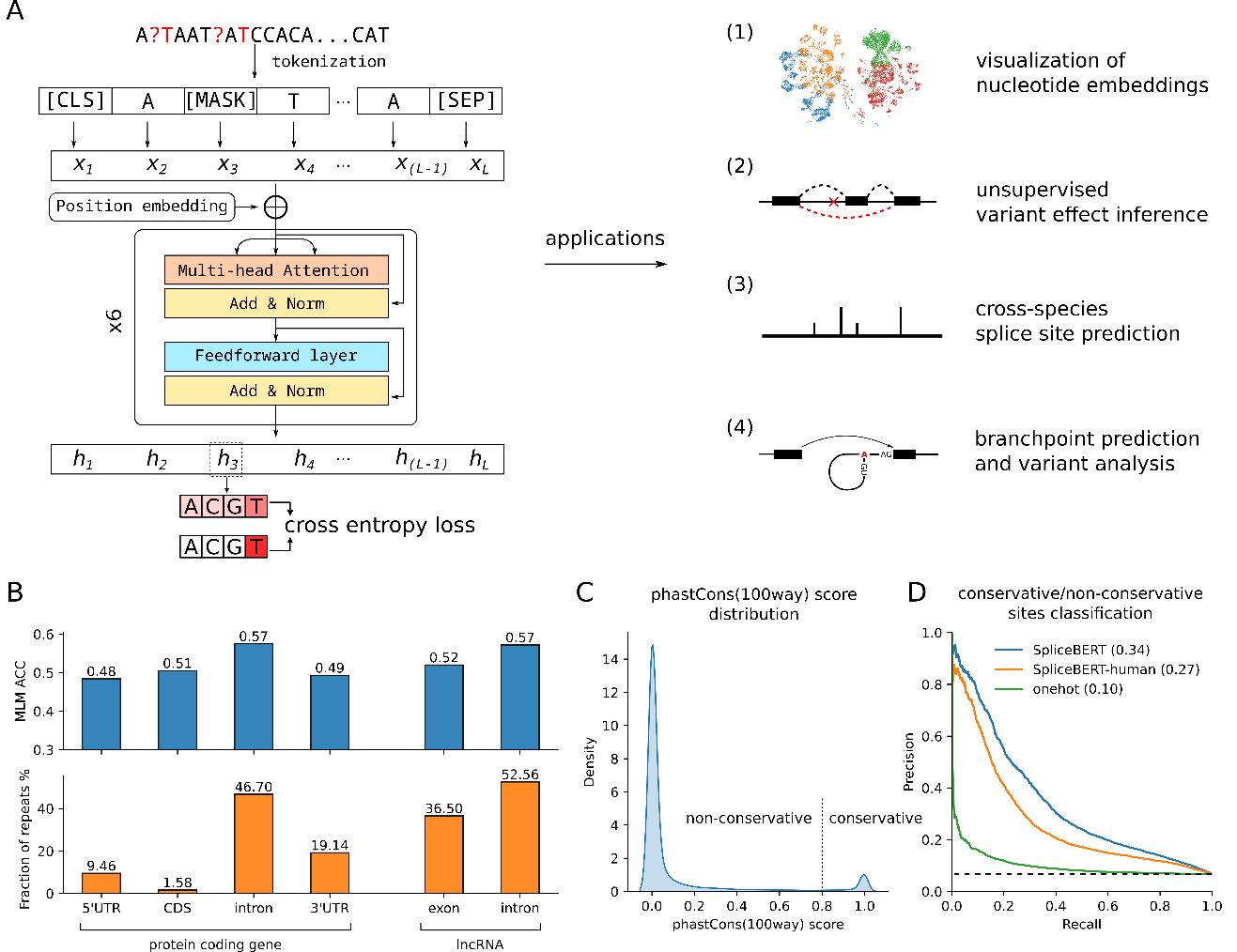
RNA splicing is an important post-transcriptional process of gene expression in eukaryotic cells. Predicting RNA splicing from primary sequences can facilitate the interpretation of genomic variants. We developed a novel self-supervised pre-trained language model, SpliceBERT, to improve sequence-based RNA splicing prediction. Pre-training on pre-mRNA sequences from vertebrates enables SpliceBERT to capture evolutionary conservation information and characterize the unique property of splice sites. SpliceBERT also improves zero-shot prediction of variant effects on splicing by considering sequence context information, and achieves superior performance for predicting branchpoint in the human genome and splice sites across species. Our study highlighted the importance of pre-training genomic language models on a diverse range of species and suggested that pre-trained language models were promising for deciphering the sequence logic of RNA splicing.