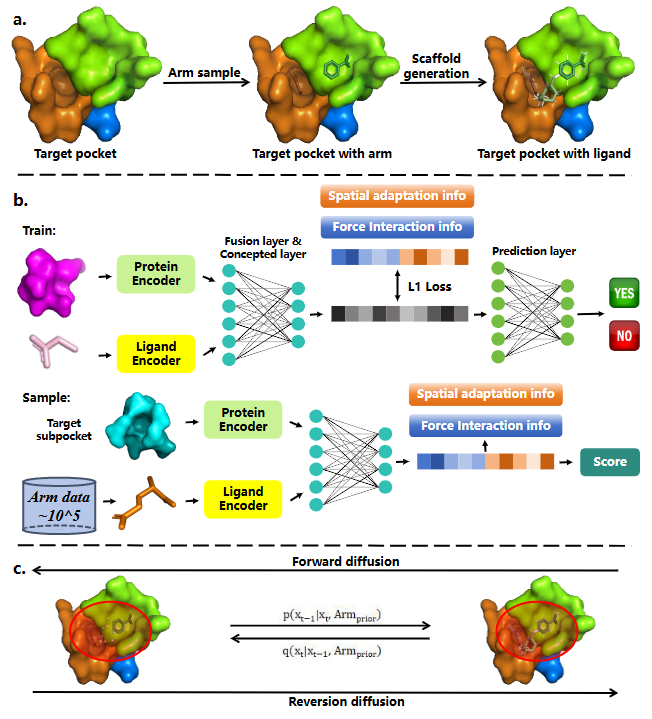
AI for Chemical Molecule
Our research focuses on advancing small-molecule modeling through deep geometric learning, multimodal large language models, and knowledge-driven molecular representations. We developed frameworks such as GIT-Mol, which unifies graph, image, and textual information for comprehensive molecular understanding, and 3D-Mol, which leverages geometric decomposition and contrastive pretraining to capture conformational structure with high fidelity. In parallel, we explore knowledge-prior and concept-injection models, integrating chemical rules, functional-group semantics, and synthesis-aware constraints directly into the representation or generation process to ensure chemically grounded outputs. Building on these capabilities, we further investigate reasoning-oriented molecular models and agent-style systems that enable stepwise molecular refinement and feedback-guided optimization. Together, these directions aim to create a unified, geometry-aware, knowledge-enhanced foundation for intelligent small-molecule modeling and design.
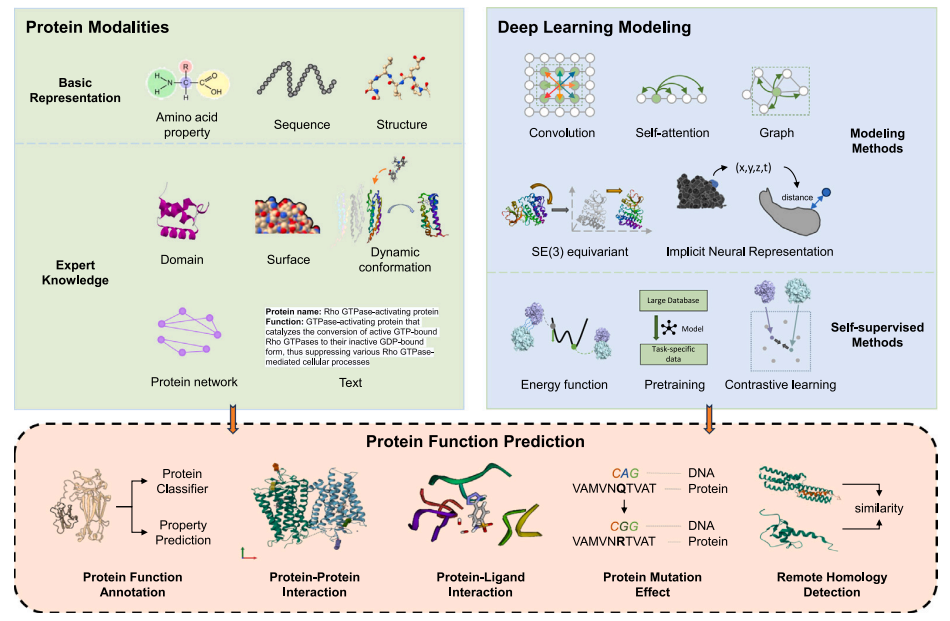
AI for Protein Science
This research theme explores AI for Protein Science, with a focus on combining geometric representation learning and multimodal large language models to advance our understanding of protein behavior. The goal is to develop unified computational frameworks that jointly capture protein geometry, sequence context, and functional signals. Our work includes enhancing sequence and structural representations by integrating protein domain information, enabling models to utilize biologically meaningful organizational units. We further investigate the decomposition of protein structures into local geometry–aware feature channels, each reflecting distinct biochemical or biophysical knowledge to build more robust and mechanism-informed models. Leveraging these enriched representations, the team develops functional prediction approaches that generalize better and remain aligned with biological principles. We also adapt state-of-the-art protein language models such as ESM-2 to downstream biological scenarios, including tasks like predicting mutation-induced protein stability changes. Together, these efforts contribute to building more capable and biologically grounded AI systems for protein science.
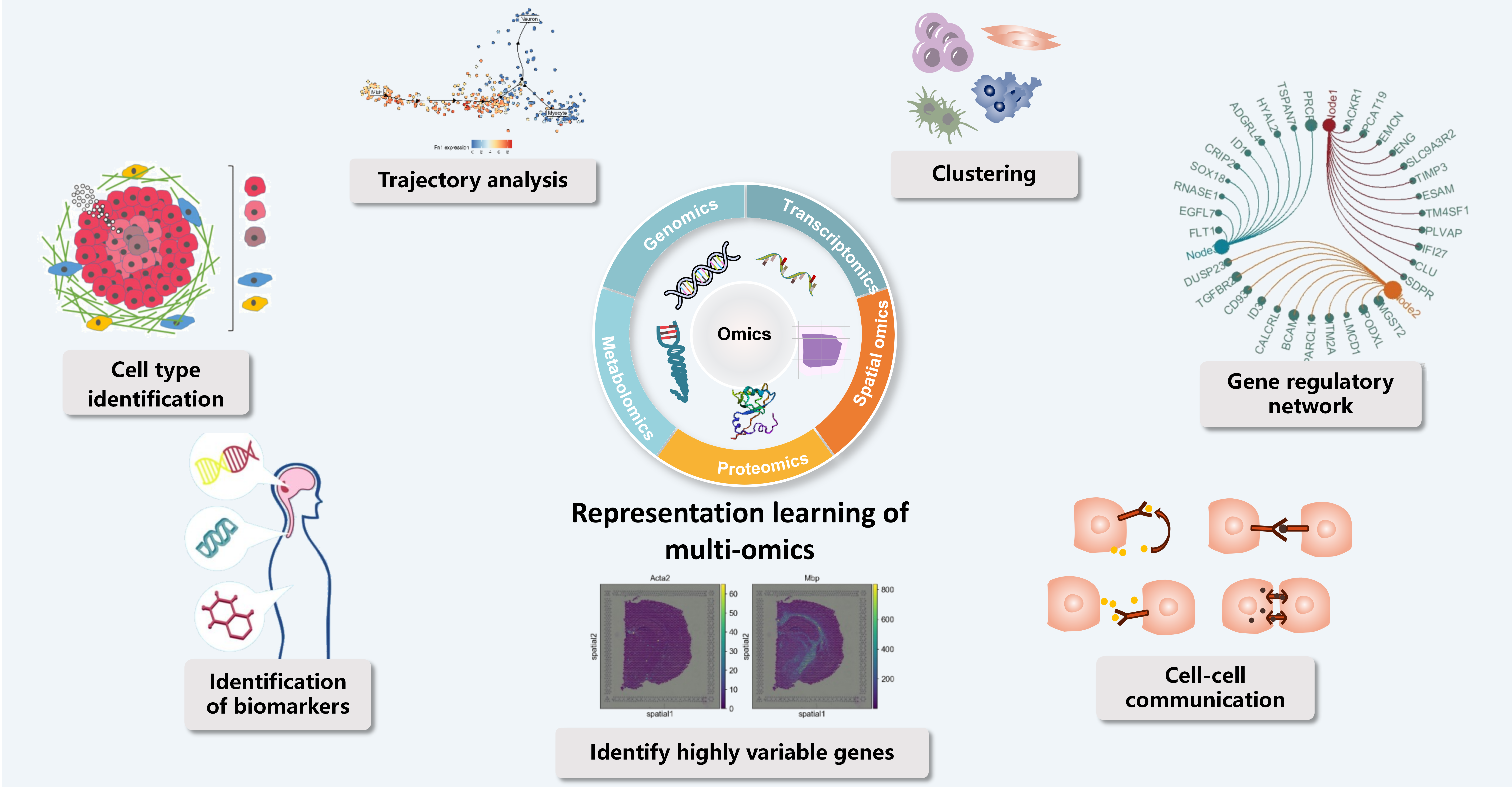
AI for Omics Data Analysis
Multi-omics modeling integrates multi-layered molecular data such as genomics and proteomics to systematically reveal cellular states, differentiation trajectories, and gene regulatory networks, providing a panoramic view for understanding fundamental biological processes like development and immunity. In traditional biology, it supports cell type identification, highly variable gene detection, and cell-cell communication analysis, thereby elucidating the dynamic regulatory mechanisms of tissue function and life activities. In oncology, multi-omics approaches enable the identification of driver mutations, biomarkers, and tumor heterogeneity, aiding in cancer subtyping, prognosis assessment, and targeted therapy design. Deep learning techniques such as representation learning and clustering analysis can extract key features from high-dimensional, sparse multi-omics data, achieving cross-modal information fusion and data imputation. Combined with technologies like spatial transcriptomics, multi-omics modeling can also dissect the tumor microenvironment at spatial scales, revealing cellular interactions and immune escape mechanisms. Despite challenges such as data integration and interpretability, multi-omics modeling has become a crucial bridge connecting basic biological discoveries with clinical medical applications. Looking ahead, with continuous advances in computational methods and interdisciplinary collaboration, multi-omics analysis will further drive the development of precision medicine and systems biology.